

Publication Highlights
|
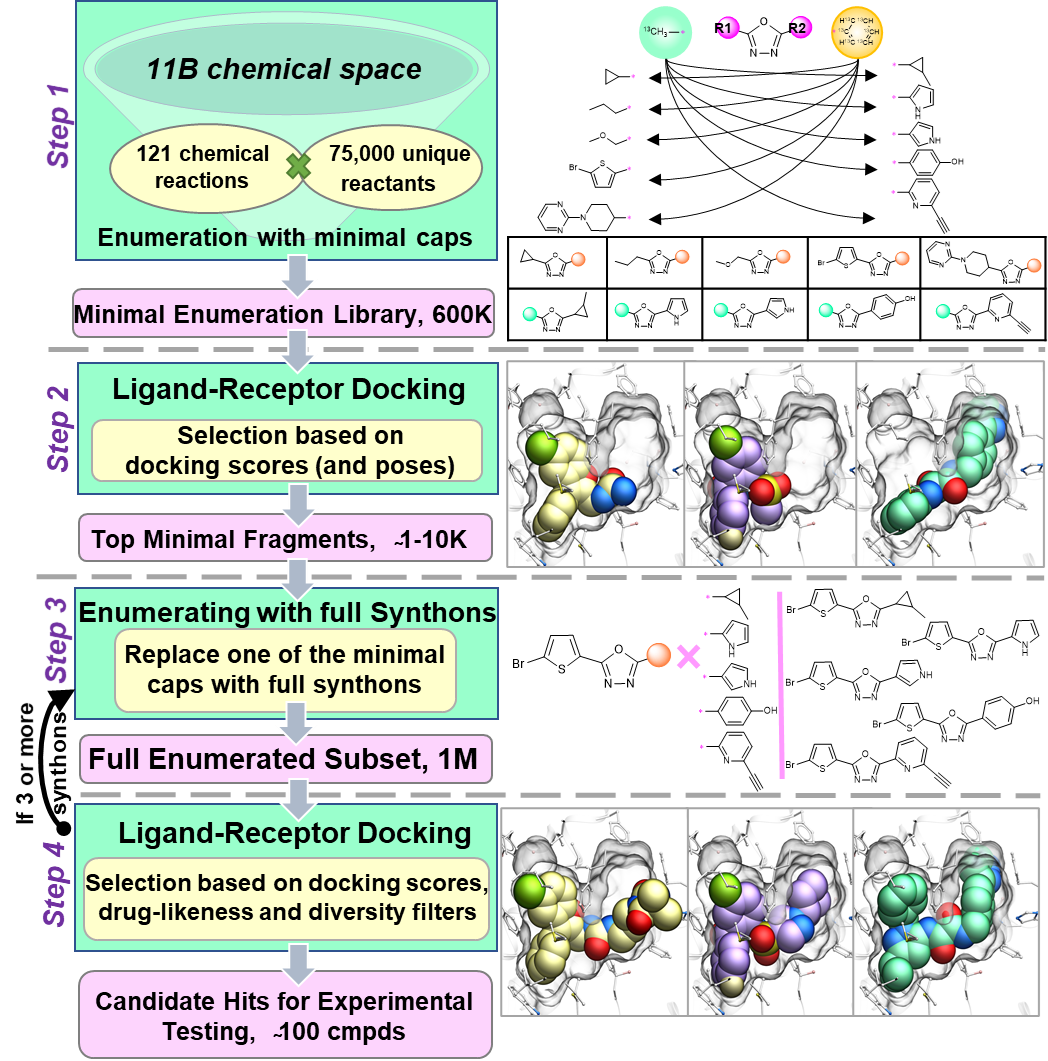
Structure-based virtual ligand screening is emerging as a key paradigm for early drug discovery owing to the availability of high-resolution target structures and ultra-large libraries of virtual compounds. However, the enormous size of virtual libraries, such as REadily AvaiLable for synthesis (REAL Space) combinatorial libraries, which already exceeds 10 billion compounds and keeps rapidly growing, now becomes a major bottleneck by itself. Here we introduce a conceptually new approach, called V-SYNTHES, to keep pace with the explosive growth of screening libraries. V-SYNTHES first identifies the best synthon-scaffold combinations as seeds suitable for further growth (see Figure), and then iteratively elaborates these seeds to select complete molecules with the best docking scores. This hierarchical combinatorial approach allows rapid detection of the best-scoring compounds in the Giga-scale chemical space while performing docking of only a small fraction (<0.1%) of the library compounds. In this study, V-SYNTHES was employed to screen REAL Space library of more than 11 billion compounds to identify new ligands of Cannabinoid receptors CB1 and CB2. The predicted by V-SYNTHES and synthesized novel compounds demonstrated 33% hit rate, including 14 sub-micromolar ligands, dramatically improving over a standard virtual screening. Selected analogs of the best hits were also synthesized, showing further improved potencies, affinities (best Ki= 0.9 nM) and CB2/CB1 selectivity (50-100 fold). V-SYNTHES was also tested on a kinase target, ROCK1, further supporting its utility for lead discovery, The approach is further scalable to Terra-scale combinatorial libraries and potentially adaptable for other docking algorithms. |
|
Melatonin receptors MT1 and MT2 are involved in synchronizing circadian rhythms and are important targets for treating sleep and mood disorders, type-2 diabetes, and cancer. We performed large scale structure-based virtual screening for new ligand chemotypes using recently solved high-resolution 3D crystal structures of agonist-bound MT receptors. Experimental testing of 62 screening candidates yielded the discovery of 10 new agonist chemotypes with sub-micromolar potency at MT receptors, with the best one reaching 0.36 nM. Six of these molecules displayed selectivity for MT2 over MT1. Moreover, the two most potent agonists had dramatically reduced arrestin recruitment at MT2, while one other compound was devoid of Gi signaling at MT1, implying biased signaling. This study reveals the utility of MT receptor structures for the structure-based discovery of selective agonists as potential leads for the treatment of insomnia, circadian rhythm, mood disorders, and type 2 diabetes. |
|
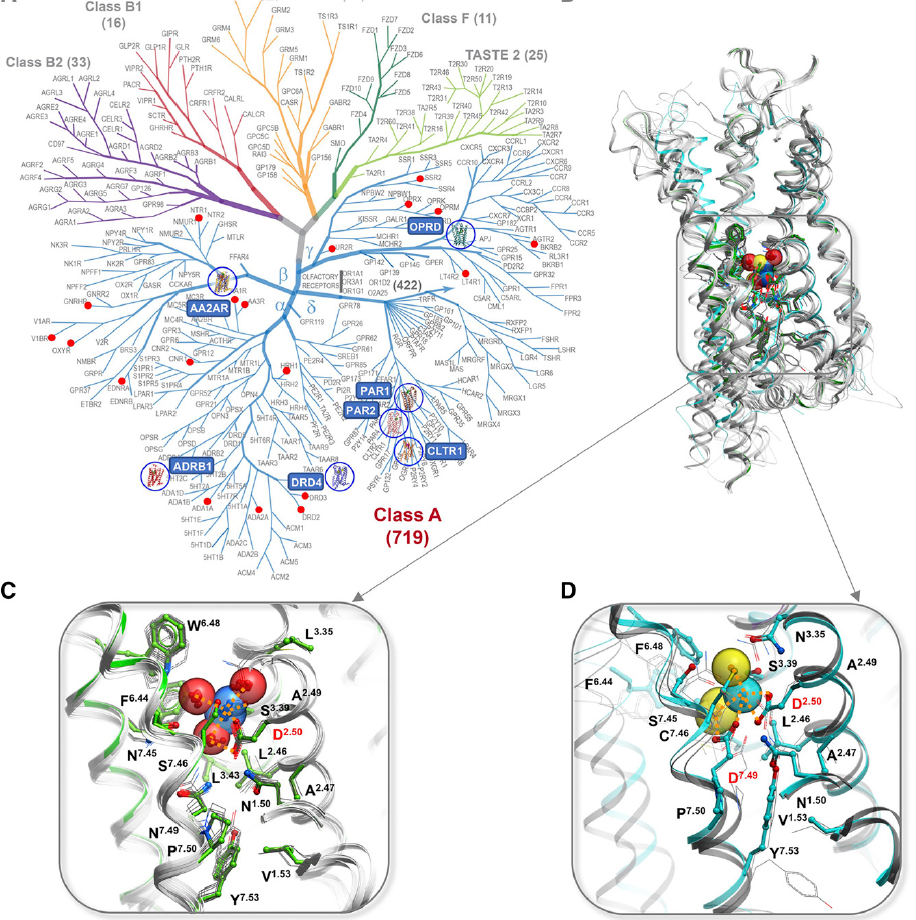
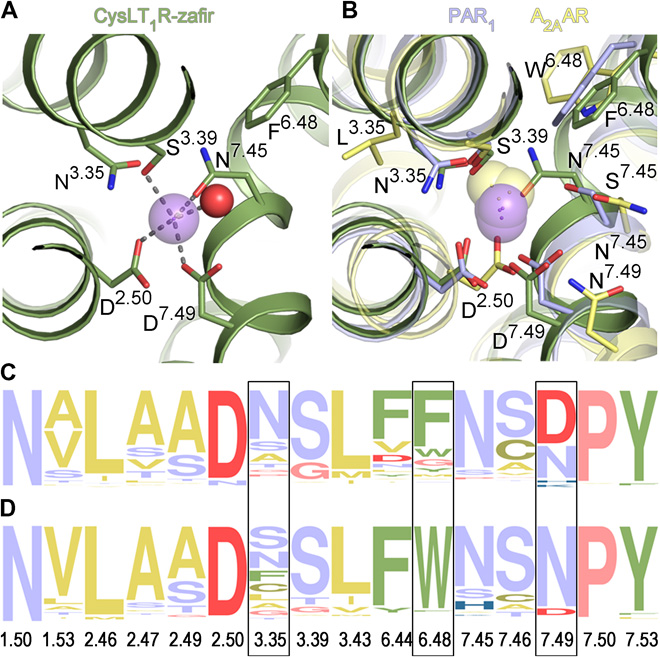
This comprehensive review describes the role of monovalent and divalent ions in GPCRs structure and function. It focuses on recent developments from our lab and others in the structural, functional, and computational characterization of the highly conserved sodium site in class A receptors first described and analyzed by Katritch lab. This involves deciphering the functional properties on sodium ion transfer across the membrane and potential coupling of sodium translocation with protonation of the key residues on the sodium path. Another key direction, described in the review is the design of new bitopic ligands targeting the sodium pocket, conferring them unusual functional properties. |
|
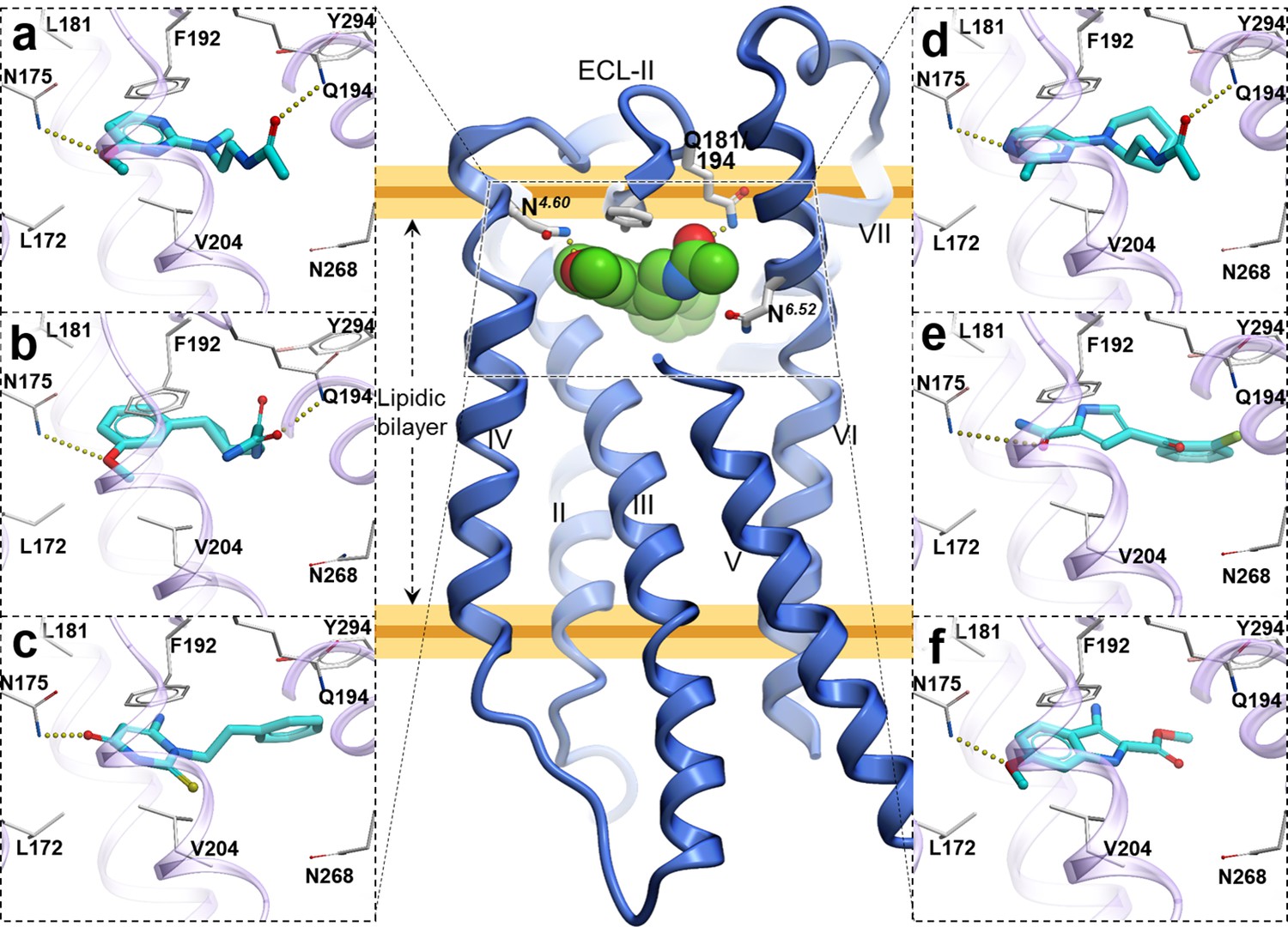
The Cysteinyl Leukotriene receptor CysLT1R mediates inflammatory processes and plays a major role in numerous disorders, including asthma, allergic rhinitis, cardiovascular disease, and cancer. To gain a deeper insight into the functional mechanisms of CysLTRs, we performed computational analysis of CysLT1R binding properties and dynamics based on the receptor structures with antagonists, solved by Dr. Cherezov lab. Comparative analysis of structural features and conformational state of the receptors, interactions of co-crystallized and computationally docked ligands, revealed a number on new properties of complexes, including lateral ligand access to the orthosteric pocket between transmembrane helices TM4 and TM5, an atypical pattern of microswitches, and a distinct four-residue–coordinated sodium site. Extensive molecular mechanics characterization also revealed a new dynamic entry mechanism for the ligands from the membrane layer. These results provide important insights and structural templates for rational discovery of safer and more effective CysLT ligands. |
|
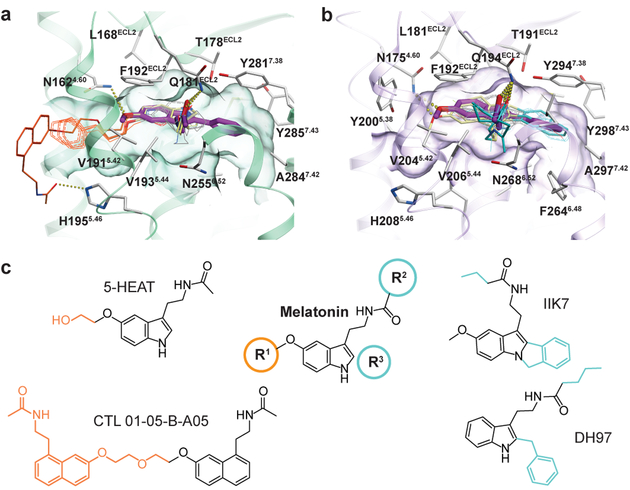
The human MT1 and MT2 melatonin receptors help to regulate circadian rhythm and sleep patterns. This paper describes the structure of the MT2 receptor in complex with 2 different agonists solved by Cherzov lab. Based on this structural information, we analyzed functional dynamics and structural basis of subtype selectivity for Melatonin receptors. The analysis reveals that, despite the conservation of the orthosteric ligand-binding site residues, there are notable conformational variations that result in different agonist binding kinetics. We also performed docking, suggesting that both a narrow side channel and the opening towards the solvent in the extracellular part of the receptor can serve as an entry path for the ligand. Moreover, we show that the side channel accommodates bulky bitopic ligands in MT1, while is too restricted for MT2, providing the basis for subtype selectivity and ligand access modes, which are essential for the design of highly selective melatonin tool compounds and therapeutic agents. |
|
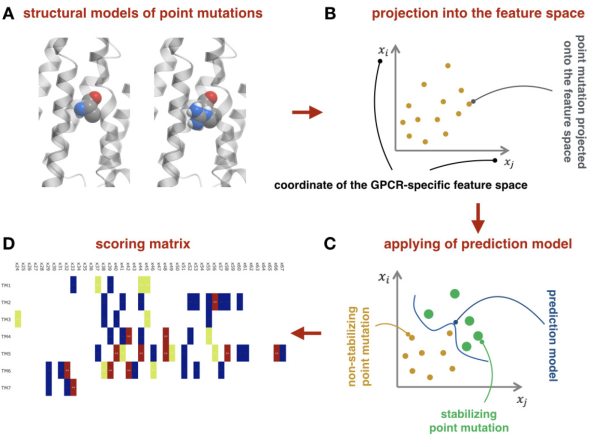
The review describes recent progress in the computational design of thermostabilizing mutations in GPCRS, focusing on our new machine-learning and structure-based methodology (CompoMug). The approach has been applied to discover stabilizing mutations for more than 20 GPCRS, and resulted in structure determination for more than 10 GPCRs of different classes, including an orphan receptor. |
|
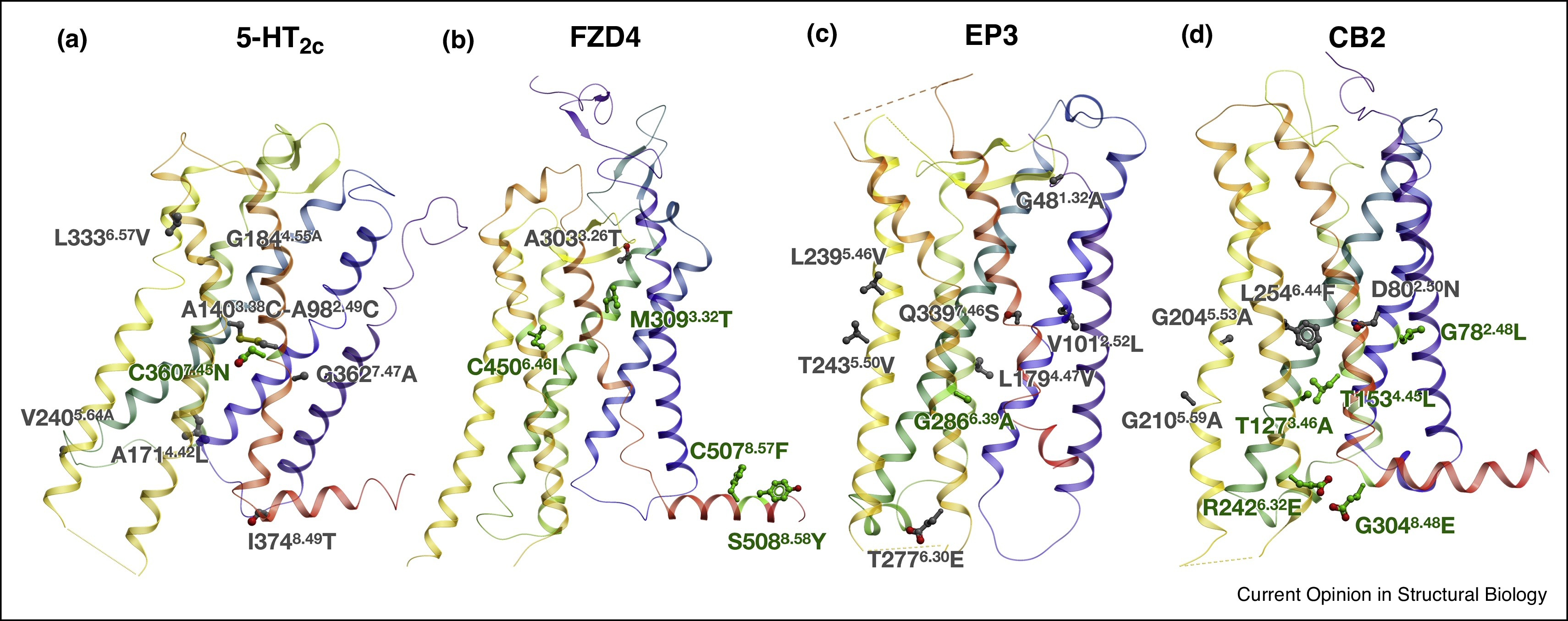
Most GPCRs must be thermostabilized for successful structure determinations and biophysical studies. Here we introduce a comprehensive computational approach to effective prediction of stabilizing mutations in GPCRs, named CompoMug, which employs sequence-based analysis, structural information, and a derived machine learning predictor. Tested experimentally on the serotonin 5-HT2C receptor target, CompoMug predictions resulted in 10 new stabilizing mutations, with an apparent thermostability gain ~8.8°C for the best single mutation and ~13°C for a triple mutant. The predicted mutations enabled crystallization and structure determination for the 5-HT2C receptor complexes in inactive and active-like states. More success stories for other GPCRs are on the way! |
|
New computational methodology, named CompoMug, is applied to predict stabilizing mutations in 5-HT2C receptor. The construct allowed structure determination of 5-HT2C in complex with agonist and with antagonist, performed by James Liu lab at iHuman, Shanghai. |
|
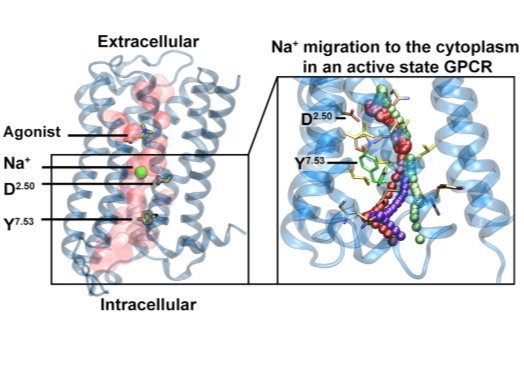
Following discovery of the conserved sodium pocket in class A GPCRs, this study analyses intracellular egress route of Na+ via umbrella sampling molecular dynamics and structural bioinformatics tools. In collaboration with Zachariae and Pisliakov groups at Dundee University, UK. |
|
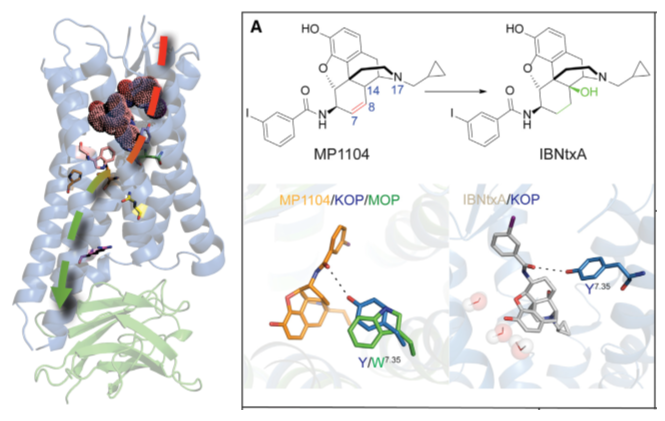
Based on the new crystal structure kappa opioid receptor (KOR), we proposed an activation mechanism in KOR and performed docking of representative agonists to the receptor, validating the structure utility in drug discovery. Roth and Wacker lab at UNC solved the structure and performed experimental binding assays. |
|
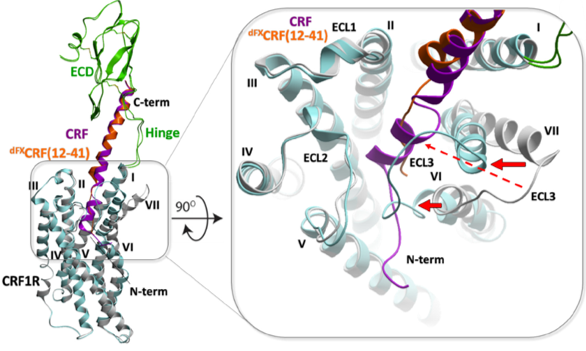
We developed a new approach to restrained energy-based conformational modeling, resulting in a dynamic structural model for the full-length complex of CRF1 receptor with peptide agonists and antagonists. We also proposed a new activation mechanism in Class B receptors, later validated by cryo-EM structures. Experimental chemical crosslinking data generated by Coin lab, Leipzig University.. |
|
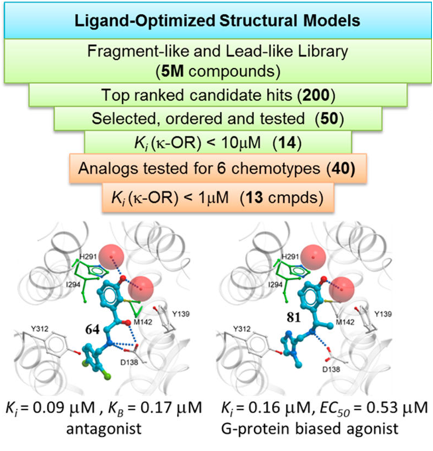
We developed virtual screening models of Kappa Opioid Receptor (KOR) and discovered six new lead like chemotypes of KOR ligands with specific functional profiles. This includes clinically important biased agonists, which selectively signal to G-protein pathways. New ligands were experimentally validated by Dr. Roth lab. |
|
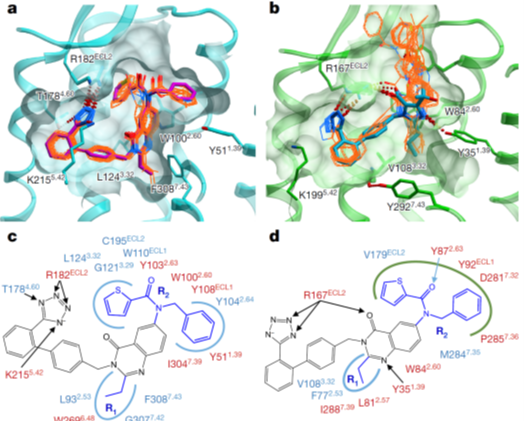
We designed fusion constructs and mutations, performed conformational modeling, ligand docking and molecular dynamics simulations to characterize new structure-function properties of Angiotensin II receptors; also proposed new mechanisms for non-canonical signaling of the type 2 receptor. Cherezov lab solved crystal structures and Merck medchem team measured ligand binding. Highlighted in Nature | News and Views |
|
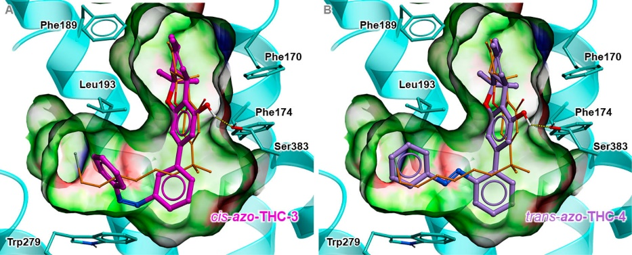
We established structural basis for cannabinoid receptor 1 binding to the new photoswitchable Δ9-THC derivatives and the mechanisms of receptor optical control. Drs. Trauner, Carriera and Frank synthesized the derivatives and characterized them in vitro and in vivo. |
|
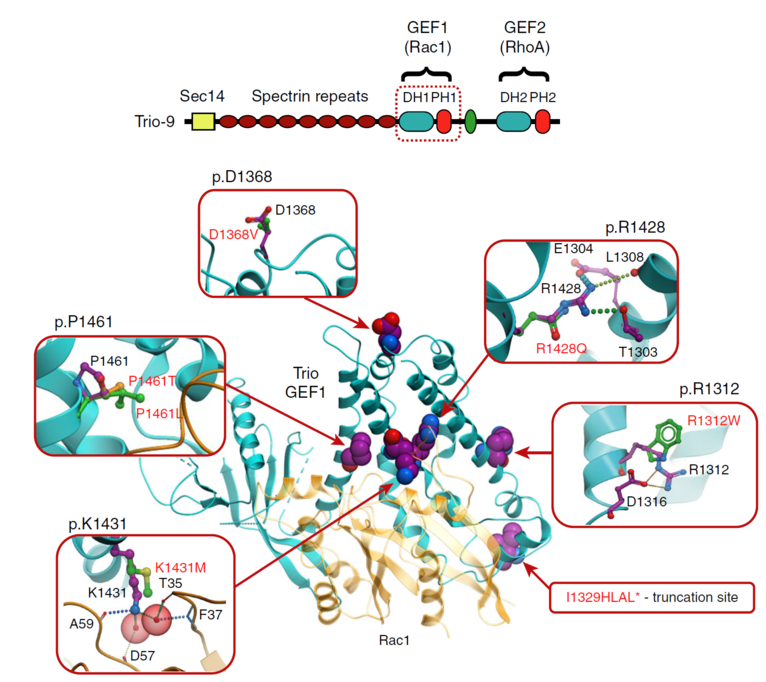
We designed statistical analysis and new structure-based modeling approach to predict effect of mutations on stability and function of Trio protein, explaining autistic phenotype of these mutants. Dr. Herring lab experimentally characterized mutation phenotypes. Highlighted in Reader's Digest as one of the “Top 12 medical breakthroughs of 2017” |
|
Structure of human delta-OR bound to the bifunctional delta-OR antagonist and mu-OR agonist tetrapeptide H-Dmt-Tic-Phe-Phe-NH2 (DIPP-NH2), solved by serial femtosecond crystallography, was used to analyze pharmacological profiles of opioid peptides, potentiating further development of improved analgesics. |

|
Despite their functional and structural diversity, G-protein-coupled receptors (GPCRs) share a common mechanism of signal transduction via conformational changes in the seven-transmembrane (7TM) helical domain. New major insights into this mechanism come from the crystallographic discoveries of a partially hydrated sodium ion that is specifically bound in the middle of the 7TM bundle of multiple class A GPCRs. This review discusses the remarkable structural conservation and distinct features of the Na+ pocket in this most populous GPCR class, as well as the conformational collapse of the pocket upon receptor activation. New insights help to explain allosteric effects of sodium on GPCR agonist binding and activation, and sodium's role as a potential co-factor in class A GPCR function. |
|
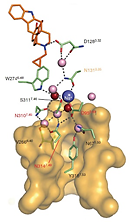
Study of allosteric sodium in GPCRs selected as a top 10 Science Signaling Breakthrough of 2014 The 1.8 Å high-resolution crystal structure of the human delta-opioid receptor (delta-OR) reveals the presence and fundamental role of a sodium ion in mediating allosteric control of receptor functional selectivity and constitutive activity. The distinctive delta-OR sodium ion site architecture is centrally located in a polar interaction network in the seven-transmembrane bundle core, with the sodium ion stabilizing a reduced agonist affinity state, and thereby modulating signal transduction. Site-directed mutagenesis and functional studies reveal that changing the allosteric sodium site residue Asn 131 to an alanine or a valine augments constitutive beta-arrestin-mediated signalling. Asp95Ala, Asn310Ala and Asn314Ala mutations transform classical delta-opioid antagonists such as naltrindole into potent beta-arrestin-biased agonists. The data establish the molecular basis for allosteric sodium ion control in opioid signalling, revealing that sodium-coordinating residues act as 'efficacy switches' at a prototypic G-protein-coupled receptor. |
|
The Smoothened receptor (SMO) mediates signal transduction in the hedgehog pathway, which is implicated in normal development and carcinogenesis. SMO antagonists can suppress the growth of some tumours; however, mutations at SMO have been found to abolish their anti-tumor effects, a phenomenon known as chemoresistance. This study reports three crystal structures of human SMO bound to the antagonists SANT1 and Anta XV, and the agonist, SAG1.5, at 2.6-2.8 Å resolution. The structural information combined with molecular modeling and rationally designed mutations reveal distinct interactions at D473(6.54f), establishing the structural basis for the differential effects of chemoresistance mutations on SMO antagonists. |
|
This review discusses how crystallography of GPCRs, combined with biochemical/biophysical techniques and molecular modeling helps to redefine our understanding of receptor-ligand recognition and functional mechanisms. Analysis of the structures is also shedding light on a structural basis of allosteric modulation and biased signaling, revealing the receptors as allosteric machines that are controlled not only by ligands but also by ions, lipids, cholesterol, and water. |

|
We performed prospective virtual screening for orthosteric and allosteric ligands of the human dopamine D3 receptor (D3R) using two optimized crystal-structure-based models: the receptor with an empty binding pocket, and the receptor complex with dopamine. Biochemical and functional characterization of predicted candidates revealed 14 novel orthosteric ligands (56% hit rate), and 8 novel allosteric ligands (32% hit rate), with compound 7 showing the highest potency of dopamine inhibition (IC50 = 7 nM). Compounds targeting the allosteric site lack the anchoring amino group and display a variety of functional activity profiles consistent with noncompetitive allosteric modulation of dopamine signaling. The high affinity and ligand efficiency of the chemically diverse hits identified in this study suggest utility of structure-based screening targeting allosteric sites of GPCRs. |
|
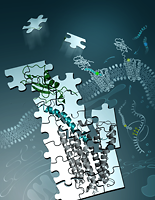
A novel hybrid approach for determining structures of key cellular receptors in complex with their peptides directly in mammalian cells was developed in Collaboration with Salk Institute Lei Wang and Irene Coin. By incorporating unnatural amino acid photochemical and new click-chemical probes into the intact receptor expressed in the native membrane of live cells, 44 intermolecular spatial constraints have been derived for the ligand-receptor interaction. Our group developed computational methodology to harness these chemical crosslinking data and to assemble the full molecular complex puzzle from multiple pieces of structural information. |

|
Binding of the glucagon peptide to the glucagon receptor (GCGR) triggers the release of glucose from the liver during fasting; thus GCGR plays an important role in glucose homeostasis. Thus study reports the crystal structure of the seven transmembrane helical domain of human GCGR at 3.4 A resolution. We used energy-based modeling and crosslinking data to compliment the crystal structure by a hybrid model of full length GCGR bound to glucagon. The hybrid model suggests that the 'stalk' region in the first transmembrane helix helps to position the extracellular domain relative to the membrane to form the glucagon-binding site that captures the peptide and facilitates the insertion of glucagon's amino terminus into the seven transmembrane domain. This model, validated with by extensive site-specific mutagenesis, provides key insight into the receptor molecular recognition and signaling by its native ligand. |

|
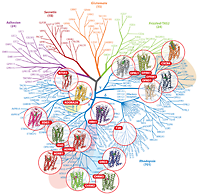
This review outlines the common mechanisms and structural diversity of GPCRs at different levels of homology, as well as the impact of the structures on drug discovery. The rapidly expanding repertoire of GPCR structures provides a solid framework for experimental and molecular modeling studies, and helps to chart a roadmap for comprehensive structural coverage of the whole superfamily and an understanding of GPCR biological and therapeutic mechanisms. |